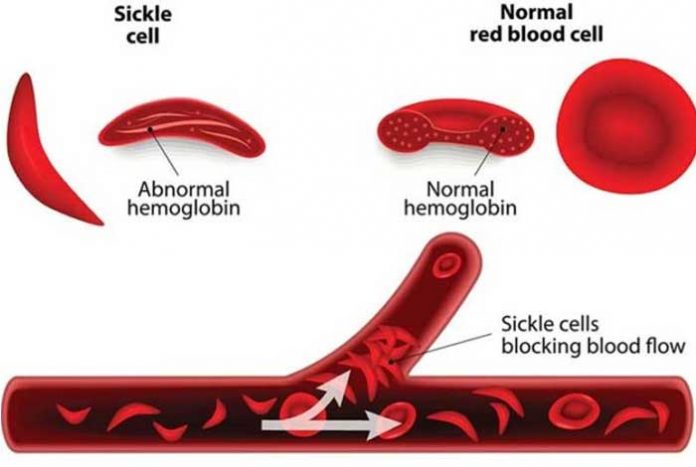
Sickle cell anemia, also known as sickle cell disease (SCD) is a genetic disease, marked by the presence of sickle-shaped red blood cells (RBCs). These defective RBCs rupture easily, leading to a medical condition known as anemia.
The red blood cells are a major component of our blood, which house an iron-containing protein, called hemoglobin. This hemoglobin, in turn, carries oxygen, which is necessary for normal functioning of our vital organs. Normally our red blood cells are disc-shaped, smooth and deformable, which move easily through the blood vessels, thus effectively supplying all the organs.
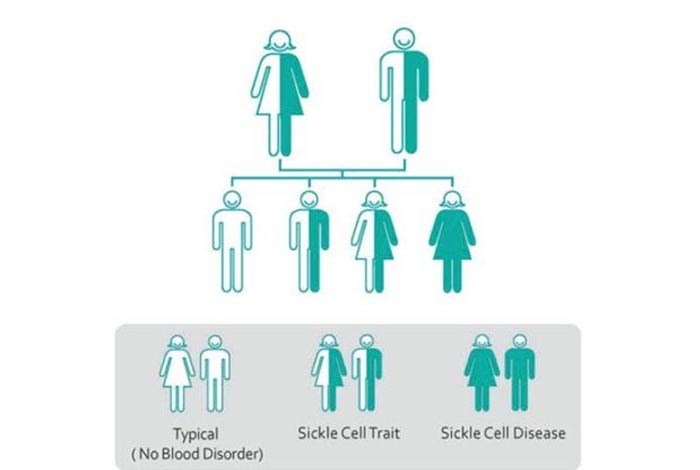
Inheritance of two sickle cell genes, one from each parent, renders the RBCs to attain a sickle shape, or a crescent shape. These deformed RBCs have a shorter life-span (10-20 days only), which means that these RBCs break down easily. An inability to compensate for the lost RBCs creates a constant deprivation of RBCs, leading to a condition known as anemia. Also, these RBCs are inflexible, posing immense trouble in passing through blood vessels, thus causing ineffective oxygen supply to all our vital organs. This hampered oxygen supply increases the risk of developing stroke, gallstones, blindness, and even organ damage.
A notable fact here is that inheritance of only a single sickle cell gene, from either parent, leads to a condition known as sickle cell trait, which is different from sickle cell disease. Sickle cell trait usually does not produce any symptom and is not a fatal condition.
It has been seen that sickle cell disease occurs more commonly in areas affected by malaria. However, people with a sickle cell trait are resistant to malaria. As per an estimate by Centers for Disease Control and Prevention, SCD affects about 1 lac Americans. SCD involves more people of black ethnicity. It affects 1 in every 365 Black or African-American births. 1 in every 13 Black or African-American births are affected by sickle cell trait.[1]
To generate awareness about this dreadful genetic disease, since 2008, 19th June is celebrated as “World Sickle Cell Day” every year.
Although fatal, sickle cell disease can be cured with a bone marrow transplant. But due to its potential life-threatening side-effects, it is rarely performed. Thus, sickle cell disease is usually managed through medications and blood transfusions.
Types
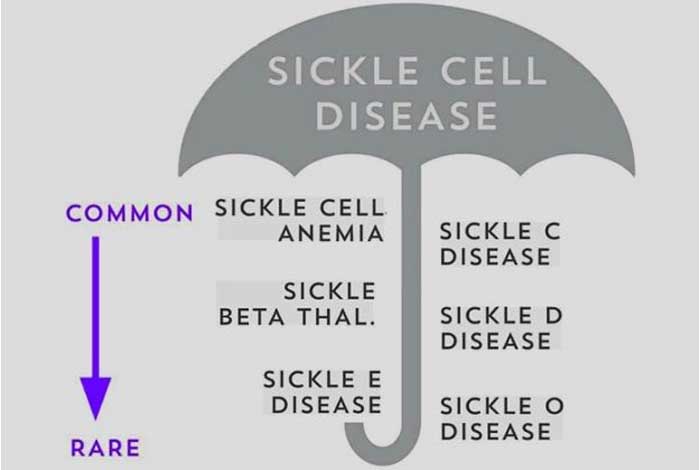 Inherited genetic mutation is the chief cause of sickle cell anemia. Based on the inheritance pattern of sickle gene, there are several types of sickle cell disease. These are described below.
Inherited genetic mutation is the chief cause of sickle cell anemia. Based on the inheritance pattern of sickle gene, there are several types of sickle cell disease. These are described below.
- Hemoglobin SS: This type of SCD occurs when the abnormal hemoglobin S gene is inherited by both the parents. It is the most common type of sickle cell disease and is generally the most severe form of sickle cell anemia.
- Hemoglobin SC: This is the second most common variant of SCD. It occurs when one of the parent transfers defective hemoglobin S gene, while the other one transfers hemoglobin C (HbC) gene. The symptoms of this condition are similar to that of Hemoglobin SS sickle cell anemia but the anemia seen in this type is milder than that of Hemoglobin SS sickle cell anemia.
- Hemoglobin Sβ+ (beta plus): Normally, hemoglobin consists of two alpha and two beta chains. Beta- thalassemia affects the production of beta globin gene. It causes reduced size of red blood cells because of minimized production of beta protein. Inheritance of beta- thalassemia gene along with that of HbS gene, produces this form of sickle cell anemia. It is less severe as compared to other forms.
- Hemoglobin Sβ0 (beta zero): This is similar to hemoglobin S-beta plus sickle cell disease, caused by inheritance of beta- thalassemia gene and Hb S gene. However, the difference lies in the severity of the disease. Hemoglobin S beta plus causes reduced beta globin production of hemoglobin, whereas hemoglobin S beta zero causes complete absence of beta globin protein. Hence, hemoglobin S beta zero is more severe than hemoglobin S beta plus.
- Other types: Besides the one mentioned above, there are several rare types of sickle cell diseases, including Hemoglobin SD, Hemoglobin SE and Hemoglobin SO.
Symptoms
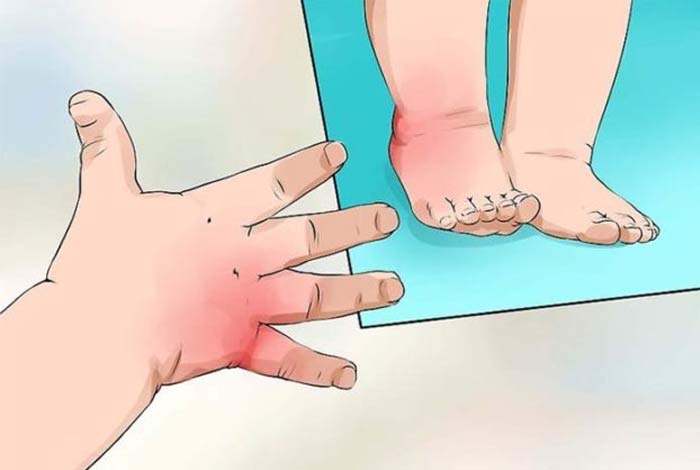
The symptoms in SCD occur due to reduced number of RBCs (medically referred to as anemia) and due to reduced oxygen supply to organs (because of inability of sickle shaped RBCs to pass easily through blood vessels). Although there are several types of SCDs, they all produce similar symptoms varying in severity. The commonly encountered symptoms are as follows.
- Anemia: As already mentioned, sickle- shaped RBCs are much more fragile than healthy disc-shaped RBCs. Hence, these sickle- shaped RBCs have a very short life-span (only 10 to 20 days), as compared to that of healthy RBCs (120 days). Hence, the RBCs in SCD rupture too frequently, causing a constant state of reduced RBCs, known as anemia.
- Excessive fatigue and weakness: Because of anemia, the vital organs receive a low oxygen supply, thus causing fatigue and weakness.
- Painful episodes: When sickle-shaped RBCs obstruct the blood vessels, they may produce painful episodes, which are also known as crisis. Some people may experience few crises per year, while others may experience many. Also, the duration of these episodes may vary from few days to weeks.
- Painful swelling of feet and hands: Obstruction of blood flow to hands and feet, may produce painful swelling.
- Frequent infections: Spleen is an organ which plays several roles, from recycling the blood cells to fighting against disease-causing bacteria, like those causing pneumonia and meningitis. SCD can damage spleen, thus causing recurrent infections.
- Retarded growth: Blood provides adequate oxygen and nutrients to the body for its optimal growth. A deprivation of hemoglobin, can impair the nutrient supply, hence delaying growth in neonates and infants. Also, a delay in puberty may occur in teenagers.
- Visual disturbances: Retina is the part of the eye that processes the visual images. However, in order to function efficiently, it requires an optimal blood flow. An impaired blood flow to retina, can occur due to sickle-shaped RBCs blocking the blood vessels. This can lead to blindness.
Other symptoms associated with SCD are as follows.
- Delayed puberty
- Gallstones, which can cause jaundice
- Bone and joint pain
- Priapism, i.e., a persistent painful erection of penis
- Ulcers or sores on the legs
- Transient Ischemic Attacks (TIAs) or strokes
- A lung condition, called acute chest syndrome, marked by fever, cough and chest pain
- Kidney problems, including blood in urine or bedwetting
Risk Factors
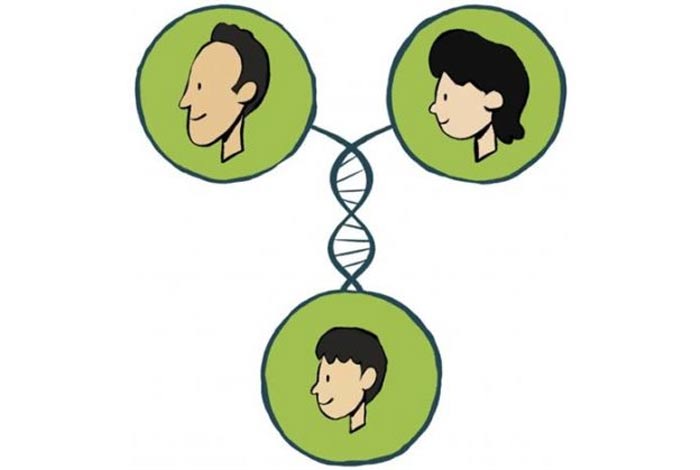
There are some classic features which put a person at a high risk of acquiring sickle cell disease. These risk factors are described below.
- Genetics: Gene responsible for causing SCD is abnormal Hemoglobin S gene. Sickle cell anemia has an autosomal recessive pattern of inheritance, i.e., for a person to develop SCD, defective gene must be acquired from both the parent. If only one parent has defective hemoglobin gene, while other parent has normal hemoglobin gene, the child won’t develop SCD, but will have sickle cell trait, which is marked by a single defective hemoglobin gene. Although such patients will have no symptoms of SCD, the patient may transmit the disease in future generations. Therefore, acquiring a pair of defective hemoglobin gene (one from each parent) is the most significant risk factor associated with SCD.
- Ethnicity: People of Black ethnicity are at the greatest risk of developing SCD. However, if a person has an ancestral origin from the Middle East, Latin America, the Mediterranean, Africa or India, the chances of acquiring SCD is high.
Do I have it?

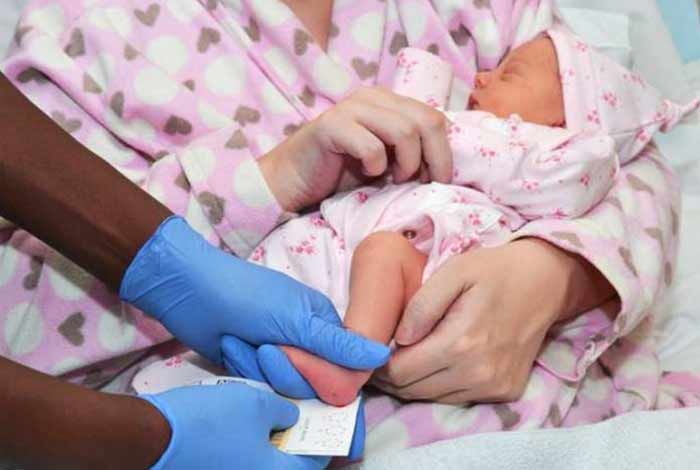
SCD is usually diagnosed at the time of birth itself during routine newborn screening. If you and your partner, both have SCD, then there is a very high chance that your child will develop it too. Being from Black ethnic origin also increases your risk of developing the disease or transmitting the disease to future generations.
Also, if you and your partner have sickle cell trait, you may be completely healthy but are considered as “carrier” of SCD. It means that there is a high chance that your child may acquire defective genes from both of you, thus putting your child at high risk of acquiring SCD.
If you are an adult and frequently experience pain in any part of body, like chest, body or joints, you may be experiencing SCD crisis, which occurs when sickle-shaped cells occlude the blood vessels.
If you also feel weak and get tired easily, you must get yourself screened for SCD. However, it must be noted that fatigue and weakness are general symptoms of any form of anemia. A thorough clinical evaluation is needed to precisely diagnose the disease.
Causes
SCD is caused by a mutation or genetic alteration in the gene that is vital to produce hemoglobin. Hemoglobin is an iron-loaded protein, which forms major component of blood. Hemoglobin has various functions. One such important function is its ability to carry oxygen from lungs to several organs and tissues of the body. Adequate oxygen supply is essential for the body to function normally.
Inheriting a pair of this defective hemoglobin gene causes the RBCs to attain a sickle shape. These deformed RBCs are rigid and sticky. It rigidness poses difficulty in passing through blood vessels, often occluding the vessels and depriving the organs of oxygen supply. The stickiness of these RBCs makes them stick to the wall of blood vessels, further hampering the blood supply of organs. This inadequate blood supply cause stroke, blindness and can severely damage the organs.
Also, these abnormal RBCs are fragile, breaking down in every 10 to 20 days. The inability of body to compensate for this loss makes the affected individual anemic.
Prevention

Being a hereditary condition, SCD can’t be prevented. However, an early recognition of the condition may prove to be of great significance in preventing the fatal complications of the disease. Although screening of newborn usually identifies the disease at its earliest stage, you may want to be prepared if you and your partner have sickle cell trait.
As per National Heart, Lung and Blood Institute, such couples should seek genetic counseling. A genetic counselor will explain you the risk factors and all the reproductive options that are available. [2]
You may also opt for genetic prenatal testing. It involves testing the parents and the fetus (developing baby in the womb) for presence of abnormal hemoglobin genes. However, this is not a method of preventing the onset of SCD but helps the parents in preparing themselves for anticipated consequences.
Diagnosis and Tests
You may seek a general physician’s consultation for your symptoms. However, on suspicion of SCD, your physician may refer you to a hematologist (a specialist of blood-related diseases).
Various methods of diagnosis are as follows.
1. Prenatal Tests: These tests are done before the baby is even born. This can be done through two processes, which are described below.
- Amniocentesis: This process involves examining the fluid surrounding the developing baby (medically known as amniotic fluid). The fluid is checked for presence of abnormal genes seen in SCD.
- Chorionic Villus Sampling: Chorionic villus is finger-like growth seen in placenta. This structure has same genetic material as is seen in the developing baby. By taking a small sample of the chorionic villus, presence of sickle gene can be checked in the baby.
2. Newborn Test: In newborns, a heel prick test is performed to remove a small amount of blood as a “spot” on a filter paper from the newborn’s heel. This blood is then analyzed to recognize abnormal hemoglobin.
3. Adult Blood Test: Adults presenting with symptoms of SCD may be asked to get a blood test for identifying abnormal hemoglobin, similar to that in babies. However, unlike babies, the blood is collected from adult’s arm veins and not by pricking heel.
Treatment
Bone marrow transplant or stem cell transplant is the only permanent cure of SCD. However, due to its potential risks, it is not usually carried out.
Thus, treatment of SCD is aimed at reducing the frequency of SCD crisis, relieving the symptoms and preventing the complications.
Various treatment methods used to treat SCD are as follows.
1. Stem cell transplant: Also known as bone marrow transplant, it involves extracting stem cells from a healthy related donor and infusing it in the affected individual through a drip. This is usually indicated in people below 16 years of age, who have severe symptoms and are not responding to any other treatment approach. Stem cells have a unique ability to transform into any cell type of the body. When infused in the affected individual, these stem cells divide and produce healthy RBCs.
However, due to some fatal side-effects associated with the procedure, it is not used as a first-line treatment.
2. Medications: Several drugs for SCD are described below.
- Pain-killers: NSAIDs may be taken to relieve the pain associated with SCD crisis.
- Antibiotics: Since SCD patients are at a higher risk of acquiring infections, they may need to be on antibiotics like penicillin on a daily basis.
- Hydroxyurea or Hydroxycarbamide: It stimulates production of normal fetal hemoglobin, which prevents formation of sickled hemoglobin. Hydroxyurea also considerably reduces the frequency of painful episodes.
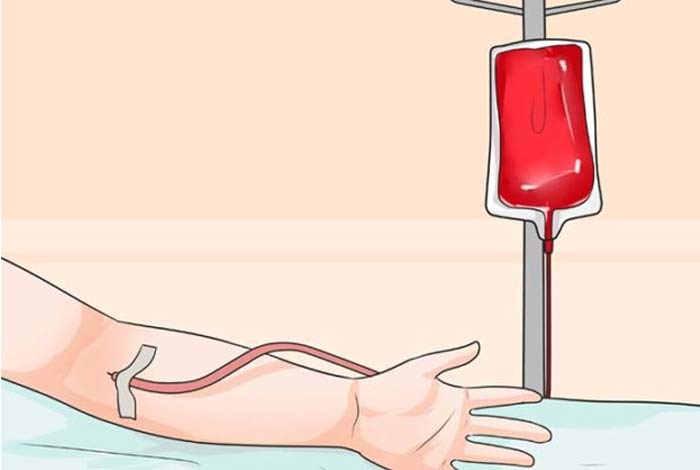
3. Blood Transfusions: If the anemia seen in SCD is persistent and severe, blood transfusions may be needed to improve the condition of the patient. However, regular use of hydroxycarbamide may reduce the need for blood transfusion. Blood transfusion or hydroxyurea is especially significant in patients with an increased risk of stroke.
4. Treatment of other symptoms: Some less frequent symptoms of SCD may be treated accordingly, as described below.
- A short-course therapy of hormonal medications may be prescribed by the doctor to induce puberty
- Gall bladder removal may be performed to treat gallstones
- Medications or drainage of blood from penis may be needed to treat priapism
- Cleaning the ulcer, followed by a dressing can help manage leg ulcers
- Acute chest treatment may require emergency treatment with antibiotics, blood transfusion and oxygen and fluid administration.
Care

SCD can be a tormenting condition for the child and also the parents. In such cases, parents should stay patient and receive appropriate medical consultation. Child with SCD may feel inferior to other children leading a normal life. Hence, it is important for parents to motivate the child and make sure that the child is leading a normal life. Teachers of affected child must be informed as these children may need more care than other healthy kids.
SCD brings along a set of hardships for everyone from little kids to adults. Painful crisis and risk of developing stroke and other complications can severely hamper the quality of life of an individual. Thus, it is important for family, friends and peer to make the patients understand that SCD is manageable through medical interventions. Providing them physical and emotional support can play a vital role in easing up the life of these individuals.
OTC
The pain-killers used in the treatment of SCD are usually available over the counter. Also, it must be noted that iron supplements are not effective in management of SCD. The use of folic acid supplements in the treatment of anemia seen in SCD is a matter of controversy.[3]
Self-Management Methods Available

SCD is a chronic, severe disorder, which invariably requires medical intervention. However, there are several self-management methods, which can complement the beneficial effects of medical therapy. These are described below.
- Know about your disease: First step to combat any disease is to know about it thoroughly. Ask your doctor about how severe your condition is, what are the treatment options and what can be the future complications of this disease. This will help you keep a check on your condition.
- Frequent Screening: If you or your child has been diagnosed with SCD, you must realize that there are several complications associated with the disease. Hence, periodic visit to the doctor is necessary to look out for complications like stroke, lipid disorders, cardiovascular conditions, etc.
- Vaccinations: Children diagnosed with SCD must receive periodic vaccinations, along with some additional vaccinations of flu and hepatitis B as these individuals are at a very high risk of acquiring infections.
- Avoid Trigger factors: There are various trigger factors that may evoke a painful crisis. These include dehydration, temperature extremes, etc. Thus, children with SCD are asked to avoid swimming and are suggested to stay in warm clothing during winters. Avoiding these trigger factors can play a significant role in managing the disease.
- Eat healthy: A diet containing fruits and vegetables can replenish the folic acid deficiency seen in SCD. Hence, a healthy diet must be taken to manage the disease.
- Exercise regularly: A regular exercise can also help in maintaining health of bones and joints in SCD. However, too strenuous exercise is not recommended. Consult your doctor before making any lifestyle change.
Natural Ways to Cure

Although medical therapy forms the mainstay of treatment of SCD, some natural methods can also aid in management of SCD. These natural methods are described below.
1. Healthy Diet: It is not uncommon to find malabsorption and nutritional deficiencies in SCD patients. Therefore, these patients are recommended to have a high caloric balanced diet, with more emphasis on vitamins, iron and folate. Leafy green vegetables, carrots, mushrooms, eggs, fishes like salmon and healthy fats, such as olive oil, coconut milk, etc.
2. Manage your stress levels: Stress can worsen the condition in SCD patients. Hence, it is essential for these individuals to manage their stress levels through gentle to moderate exercise. It must be noted that exercise should be combined with plenty of rest and sleep in order to manage SCD.
3. Do not overexert: SCD patients are already dealing with low-oxygen supply due to a lack of healthy RBCs. Overexertion can further reduce the oxygen levels, severely hampering the general well-being of an individual. Thus, too much of physical exertion can aggravate SCD crisis. Hence, avoid stressful situations.
4. Have plenty of water: Dehydration is a major trigger factor for SCD. Hence, SCD patients must make sure that they are constantly hydrated.
5. Managing pain naturally: Long-term use of painkillers can produce some disturbing side-effects. There are many natural ways to manage joint pain and muscle stiffness associated with SCD. These are as follows.
- Apply warm compresses to the affected area for 15 minutes twice or thrice a day. Warm baths or sitting in hot tubs or jacuzzi can also serve the purpose and reduce pain.
- Massage therapy has shown to reduce pain associated with SCD.
- Stretching and flexing under the supervision of a physiotherapist can also produce desirable results in managing pain.
6. Essential oils: It is a well-known fact that essential oils not just alleviate pain, but also imparts relaxation and boosts immunity. Application of peppermint oils, lavender oil and citrus oils have been seen to be effective in SCD patients.
Health Tip by Experts
Every day is a constant struggle for sickle cell anemia patients. Everyone from little kids to grown-up adults, may lose their self-esteem and feel alienated because of this condition. However, remember that with constant motivation and perseverance, you can definitely push through tough times.

")


